There have been no therapeutic clinical trials conducted with Zemimet SR 50/1000 mg tablets, however bioequivalence of Zemimet SR 50/1000 mg with co-administered gemigliptin and metformin has been demonstrated.
Gemigliptin: Summary of the safety profile: There were 1468 patients with type 2 diabetes, including 1080 patients treated with gemigliptin, and 821 patients were treated with gemigliptin 50 mg, randomized in 5 double-blind and 1 open-label, controlled clinical safety and efficacy studies, conducted to evaluate the effects of gemigliptin on glycemic control.
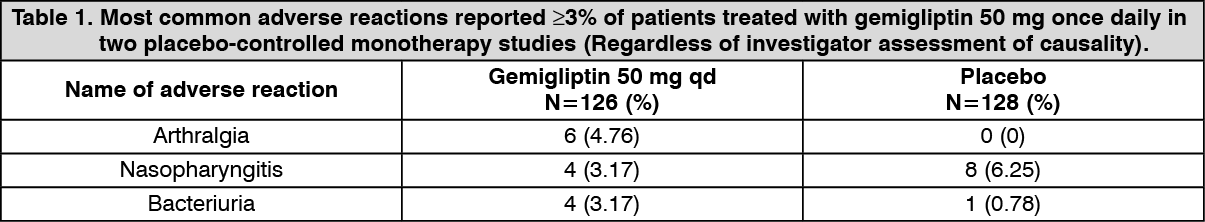
Two placebo-controlled monotherapy studies, one of 12- and one of 24-week duration, included patients treated with gemigliptin 50 mg once daily. Table 1 summarizes the most common (≥ 3% of patients) adverse reactions reported in the group treated with gemigliptin 50 mg once daily. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The 24-week monotherapy study was extended through 52-week. The adverse events that increased more than 1% during the latter 28 weeks when compared with the first 24 weeks, regardless of assessment of causality, were nasopharyngitis (4.44% vs 6.1%), upper respiratory tract infection (1.1% vs 6.1%) and increase in blood creatine phosphokinase (2.22% vs 4.88%). No new adverse events were reported in more than 2 patients (2.44%) in latter 28 weeks.
One active-controlled add-on combination therapy study with metformin included patients treated with gemigliptin 25 mg twice daily, gemigliptin 50 mg once daily and sitagliptin 100 mg once daily. Table 2 summarizes the most common (≥3% of patients) adverse reactions reported in this study. (See Table 2.)
Click on icon to see table/diagram/image
The 24-week add-on combination therapy was extended through 52-week with gemigliptin 50 mg once daily added on to stable dose of metformin. The adverse events that increased more than 1% during the latter 28 weeks when compared with the first 24 weeks, regardless of assessment of causality, were diarrhea (0.71% vs 2.7%), urinary tract infection (0.71% vs 1.8%), hypoglycemia (0.71% vs 2.7%), dizziness (0.71% vs 3.6%) and nausea (1.43% vs 2.7%). The adverse events reported in more than 2 patients (1.8%) during the latter 28 weeks were asthenia (1.8%) and myalgia (1.8%).
No clinically meaningful changes in vital signs or in ECG (including in QTc interval) were observed in patients treated with gemigliptin.
Gemigliptin 50 mg was added in patients inadequately controlled on their maximal tolerated dose of metformin and a glimepiride for 24 weeks. Table 3 summarizes the most common (≥3% of patients) adverse reactions reported in this study. (See Table 3.)
Click on icon to see table/diagram/image
In a 24 weeks clinical trial that studied initial combination with metformin, gemigliptin 50 mg and metformin were individually administered and co-administered once daily. Table 4 summarizes the most common (≥3% of patients) adverse reactions reported in this study. (See Table 4.)
Click on icon to see table/diagram/image
A 12 weeks Phase 3b clinical trial was conducted with administration of gemigliptin 50 mg in active-controlled, randomized, open-label and parallel group design in order to evaluate MAGE (mean amplitude of glycemic excursions) and safety of gemigliptin and metformin initial combination therapy to the standard combination therapy (sitagliptin and metformin, glimepiride and metformin). Table 5 summarizes the most common (≥3% of patients) adverse reactions reported in this study. (See Table 5.)
Click on icon to see table/diagram/image
The pooled safety analysis was performed in the subjects who had received gemigliptin at least once in above 6 randomized, controlled clinical studies.
The overall incidence of adverse events in patients treated with gemigliptin was similar to placebo and active-control group.
Discontinuation of therapy due to adverse events was similar in patients who received gemigliptin as compared to placebo (1.4% as compared to 0.8%).
Across all the clinical studies, there was no serious adverse event (SAE) related to gemigliptin.
Tabulated list of adverse reactions: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Table 6 presents adverse reactions which have been reported during 6 randomized, controlled clinical studies.
The adverse reactions are listed by SOC (system organ class) and PT (preferred term) with frequency. Frequencies are defined as Very common (≥1/10), Common (≥1/100 to <1/10), Uncommon (≥1/1,000 to <1/100), Rare (≥1/10,000 to <1,000), not known (cannot be estimated from the available data). (See Table 6.)
Click on icon to see table/diagram/image
Description of selected adverse reactions: Hypoglycemia: In six randomized controlled studies of gemigliptin, 15 patients (1.4%) reported hypoglycemia. The hypoglycemia experienced by patients in clinical trials was considered mostly of mild in intensity and patients fully recovered.
Hypersensitivity: In the active-controlled add-on combination study (gemigliptin as add-on to metformin therapy), two patients (1.71%) receiving 25 mg gemigliptin twice daily on a stable dose of metformin in the first 24-weeks and 50 mg once daily in the latter 28 weeks reported anaphylactic reactions, which was not related to gemigliptin (see Contraindications and Precautions).
Metformin: Table 7 presents adverse reactions which have been reported during clinical studies and in postmarketing experience. The adverse reactions are listed by system organ class and by frequency category. Frequencies are defined as very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000), not known (cannot be estimated from the available data). (See Table 7.)
Click on icon to see table/diagram/image
Gemigliptin 50 mg/Metformin 1000 mg extended release: Summary of the safety profile: There were 182 patients with type 2 diabetes treated with a single concomitant dose of gemigliptin 50 mg and metformin 1000 mg extended release (2 tablets of metformin 500 mg extended release) in 2 double-blind and 1 open-label, controlled clinical safety and efficacy studies, conducted to evaluate the effects of gemigliptin on glycemic control.
The pooled safety analysis was performed in subjects who had received a single concomitant dose of gemigliptin 50 mg and metformin 1000 mg extended release in above 3 randomized, controlled clinical studies (one as an extension of an add-on combination to metformin, one as an initial combination with metformin, and the other as a phase 3b trial evaluating MAGE).
The overall incidence of adverse events in patients treated with gemigliptin 50 mg and metformin 1000 mg extended release was 49.5% (90/182). Among them, the incidence of adverse drug reactions was 7.7% (14/182).
Across all the clinical studies, there was no serious adverse drug reactions.
Tabulated list of adverse reactions: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Table 8 presents adverse reactions which have been reported during 3 randomized, controlled clinical studies.
The adverse reactions are listed by SOC and PT with frequency. Frequencies are defined as Very common (≥1/10), Common (≥1/100 to <1/10), Uncommon (≥1/1,000 to <1/100), Rare (≥1/10,000 to <1,000), not known (cannot be estimated from the available data). (See Table 8.)
Click on icon to see table/diagram/image
Description of selected adverse reactions: Hypoglycemia: In three randomized controlled studies of gemigliptin 50 mg and metformin 1000 mg, 4 patients (2.2%) reported hypoglycemia. Hypoglycemia unawareness was reported in 1 patient (0.5%). The hypoglycemia experienced by patients in clinical trials was considered mostly of mild in intensity and patients fully recovered.
Hypersensitivity: One patient (0.5%) receiving gemigliptin 50 mg and metformin 1000 mg extended release reported hypersensitivity, which was not related to gemigliptin.
In addition, two bioequivalence studies, one was conducted under fed conditions and the other was conducted under fasting conditions, included healthy volunteers administered a single dose of a fixed-dose combination of gemigliptin/metformin 50/1000 mg (Zemimet SR 50/1000 mg) compared to a single concomitant dose of gemigliptin 50 mg and metformin 1000 mg extended release. In the first bioequivalence study conducted in healthy male volunteers under fed conditions, 10 adverse events were reported in 8 out of 24 subjects. The incidence of adverse events after receiving a single dose of Zemimet SR 50/1000 mg was 8.33% for rhinorrhea, and 4.17% for alanine aminotransferase increase, aspartate aminotransferase increased, neutrophil count decreased, and upper abdominal discomfort, respectively. The incidence of adverse events after receiving a single concomitant dose of gemigliptin 50 mg and metformin 1000 mg extended release was 4.17% for neutrophil count decreased, creatine kinase increased, dizziness, and corneal abrasion, respectively.
In a second bioequivalence study conducted in healthy volunteers under fasting conditions, 8 adverse events were reported in 7 out of 37 subjects after receiving a single dose of Zemimet SR 50/1000 mg and 11 post-dose adverse events were reported in 9 out of 37 subjects after receiving a single concomitant dose of gemigliptin 50 mg and metformin 1000 mg extended release. The most reported adverse event post-dose was asymptomatic hypoglycemia which occurred 10.81% and 21.62% after receiving a single dose of Zemimet SR 50/1000 mg and a single concomitant dose of gemigliptin 50 mg and metformin 1000 mg extended release, respectively. Other reported adverse events after receiving a single dose of Zemimet SR 50/1000 mg were hemoglobin and hematocrit decreased (2.70%), diarrhea (2.70%), loose stool (2.70%) and AST and ALT increased (2.70%), and other reported adverse events after receiving a single concomitant dose of gemigliptin 50 mg and metformin 1000 mg extended release were abnormal ECG (2.70%), nausea (2.70%), and dizziness (2.70%).
In a randomized, open-label, single oral dose, one-treatment, two-period, two-sequence, crossover food-effect bioavailability study under fed and fasting conditions in healthy Thai volunteers, 40 post-dose adverse events were reported in 19 out of 26 subjects receiving Zemimet SR 50/1000 mg. The most reported adverse event after single dose administration of Zemimet SR 50/1000 mg was asymptomatic hypoglycemia which occurred 55.00% and 60.00% under fasting and fed conditions, respectively. Other reported adverse events after single dose administration of Zemimet SR 50/1000 mg under fasting conditions were vomiting (15.00%), dizziness (10.00%), nausea (5.00%), WBC count increased (5.00%), hyperglycemia (5.00%) and symptomatic hypoglycemia (5.00%). Other reported adverse events after single dose administration of Zemimet SR 50/1000 mg under fed conditions were hemoglobin and hematocrit decreased (15.00%), dizziness (10.00%), diarrhea (5.00%), nausea (5.00%) and faintness (5.00%).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image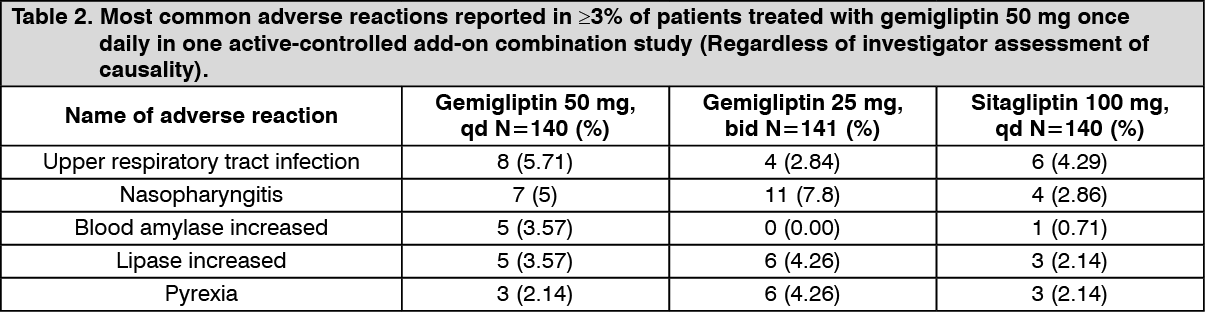 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image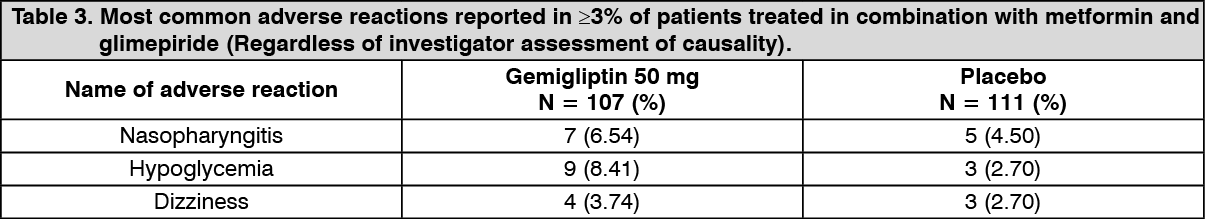 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image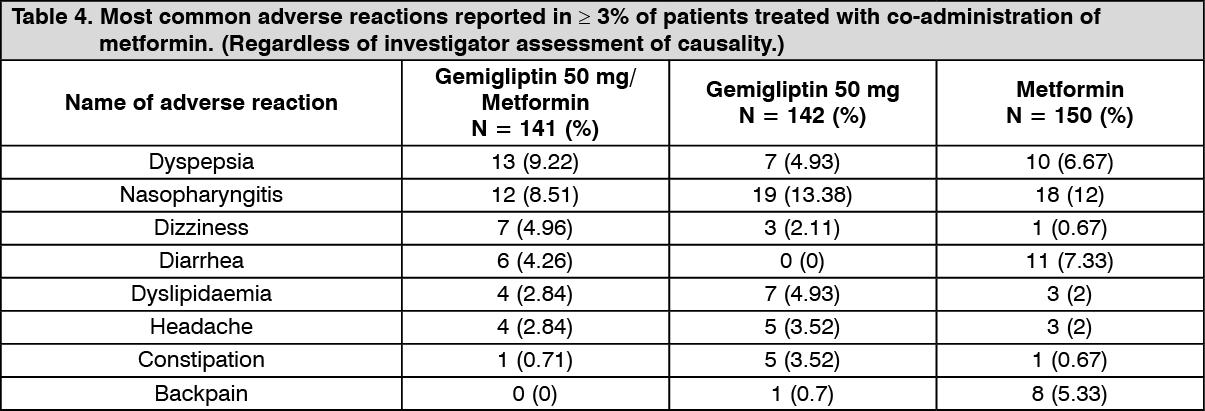 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image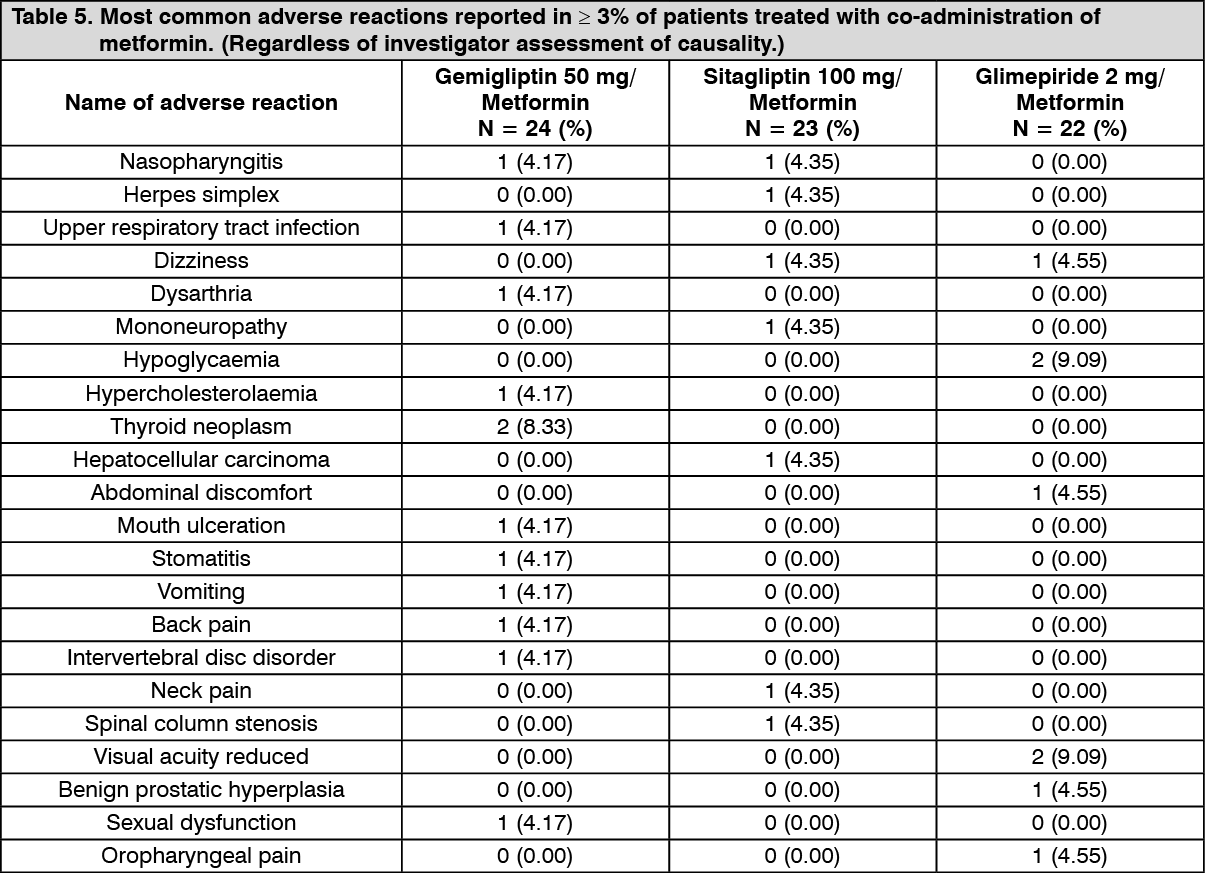 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image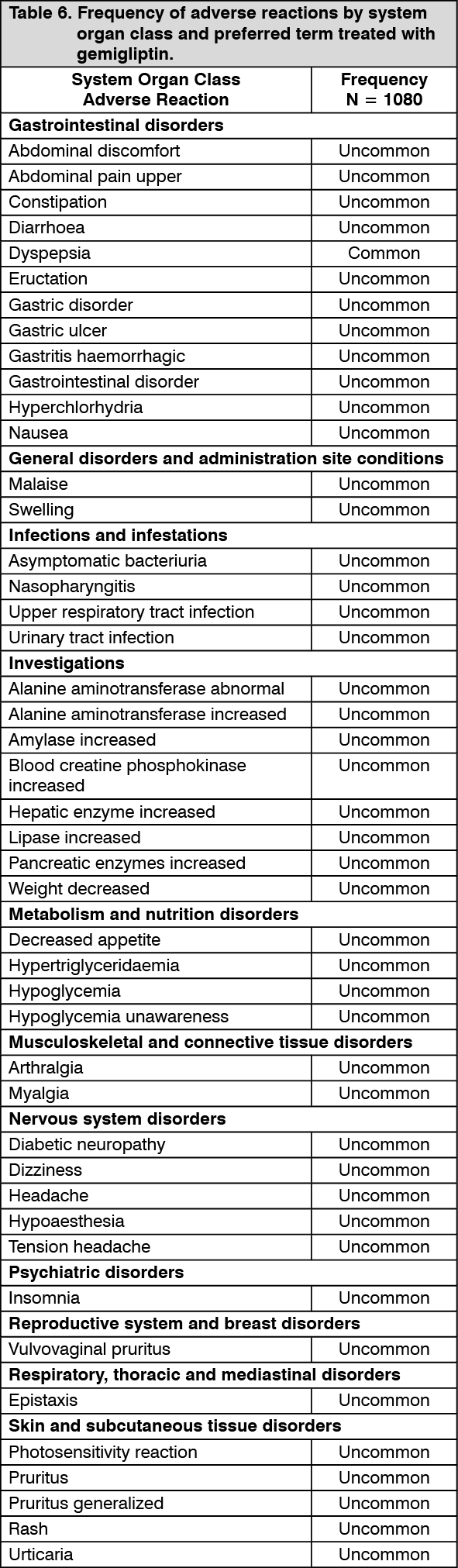 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image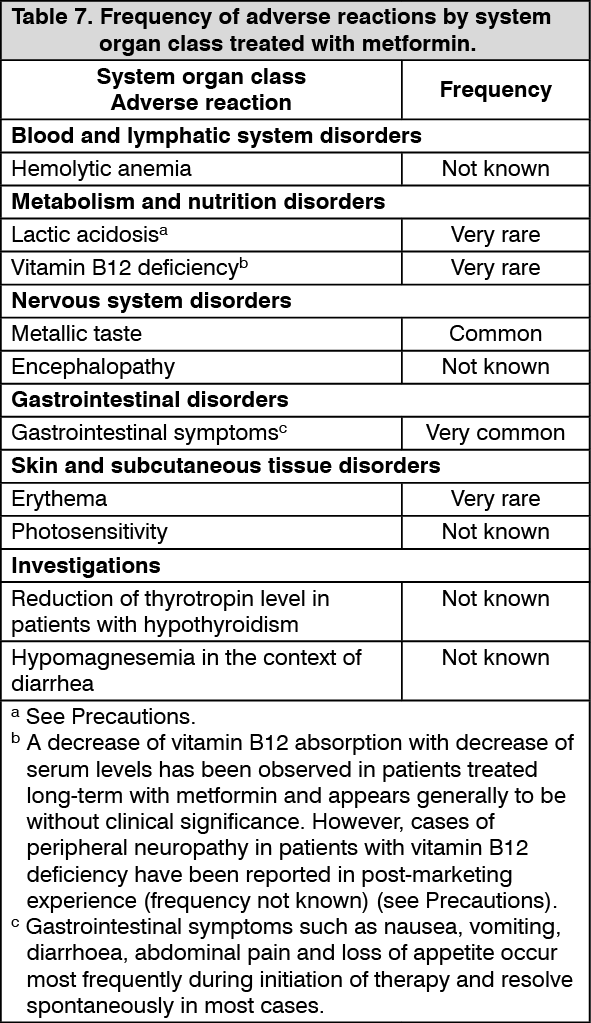 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image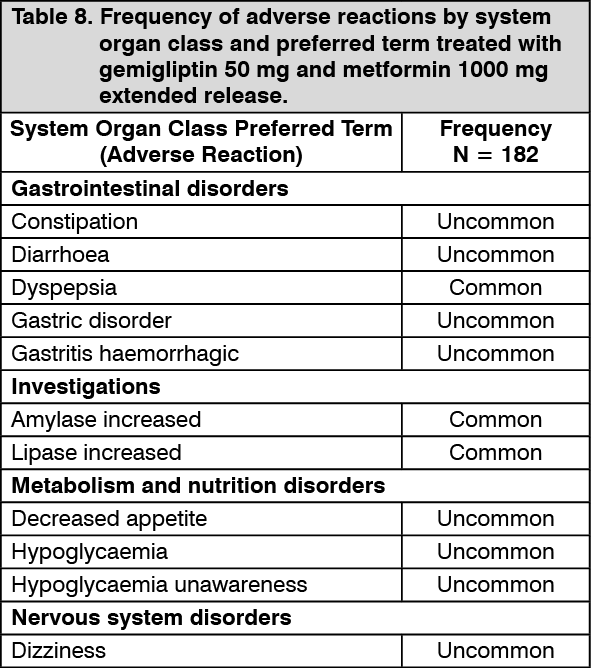 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out